Что такое синдром Гурлер
Синдром Гурлер, известный также как болезнь Пфаундлера-Гурлер, — это мукополисахаридоз I типа (МПС-I H), одна из многих форм заболеваний обмена веществ. В 1919 году его впервые описала австрийский педиатр Гертруда Гурлер.
Мукополисахаридоз I типа (МПС I) — наследственная лизосомная болезнь накопления, вызванная полным или частичным дефицитом фермента альфа-L-идуронидазы. Лизосомы в нашем организме — это «перерабатывающий центр» клетки, в котором содержатся ферменты для расщепления продуктов обмена веществ. У здорового человека альфа-L-идуронидаза работает как «утилизатор», разрушая особые полисахариды — гликозаминогликаны. Когда этого фермента не хватает, лишние вещества накапливаются, и со временем перегруженные клетки напоминают «переполненный резервуар». Так они уже не могут работать правильно.
Поскольку гликозаминогликаны участвуют в работе многих органов и систем, эта генетическая поломка оказывает влияние на весь организм в целом. Поэтому клинические проявления могут быть разными и затрагивают все органы и системы. Они проявляются задержкой роста, интеллектуальными особенностями, поражением нервной системы, сердечно-легочными нарушениями, увеличением печени и селезенкии (гепатоспленомегалией), множественными аномалиями развития скелета (дизостозами), помутнением роговицы.

Причины и механизм развития болезни
Медицинский координатор программы Ассистент Здоровья фонда «Правмир», врач общей практики, педиатр и эндокринолог Екатерина Шлимакова:
«Причиной появления МПС I типа становится мутация в гене, который отвечает за лизосомный фермент альфа-L-идуронидазу. Тип наследования: аутосомно-рецессивный. Ген IDUA, кодирующий альфа-L-идуронидазу локализован в хромосомной области 4p16.3»
Когда ген IDUA не работает, фермент не производится, гликозаминогликаны начинают бесконтрольно накапливаться. Без своевременного расщепления они превращаются в «клеточный мусор», который блокирует правильную работу соединительной ткани, сердца, органов зрения и нервной системы. Современным генетикам удалось обнаружить десятки различных мутаций в этом гене, и именно комбинация того, какой именно вариант мутации унаследовал ребенок, определяет тяжесть заболевания.
Заподозрить синдром Гурлер до зачатия нельзя, а спектр генетических заболеваний настолько широк, что возможность исключить каждое из них у пары сводится практически к нулю. При МПС I типа болезнь проявится только в том редком случае, когда оба родителя окажутся носителями пораженного гена, что само по себе встречается нечасто. При этом сами они не болеют, у них «в роду» нет семейной истории этого заболевания и, соответственно, никаких опасений, что у ребенка может проявиться синдром Гурлер. Только при планировании следующей беременности, если в семье уже появился малыш с мукополисахаридозом, родителей направляют на генетическое обследование.
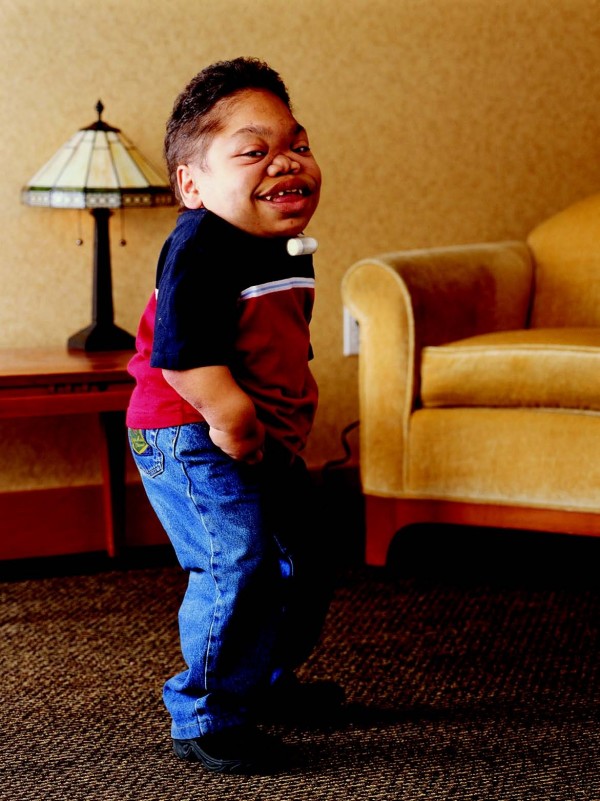
Авторские права ©2010 Valayannopoulos et al; лицензиат BioMed Central Ltd.
Три формы мукополисахаридоза I типа
МПС I типа может проявляться по-разному у каждого человека. Конкретные симптомы зависят и от так называемого «возраста дебюта», когда болезнь впервые начала проявлять себя, и от скорости прогрессирования. Симптомы не всегда нарастают стремительно.
Заболевание встречается нечасто, всего у 1 из 100 000 новорожденных. Но приблизительно у 50-80% всех пациентов оно проявляется в тяжелой форме.
При самой сложной форме, синдроме Гурлер, первые проявления можно заметить уже на первом году жизни. Страдают сердечно-сосудистая, дыхательная и нервная системы. В возрасте от 6 до 12 месяцев родители замечают у ребенка утрату приобретенных навыков и часто именно тогда впервые обращаются к врачу. В более раннем возрасте неврологи и врачи общей практики могут видеть и другие косвенные признаки, например, тугоподвижность суставов. Но это не специфический маркер именно синдрома Гурлер, и поставить такой диагноз можно только после полноценного обследования.
Синдром Гурлер-Шейе (промежуточная форма) — это форма, при которой фермент частично сохраняет свою активность, что позволяет сдвинуть дебют заболевания на более поздний возраст. В этом случае тревожные признаки могут проявиться, когда ребенку от 3 до 8 лет. При этой форме могут отсутствовать интеллектуальные нарушения, но наблюдаются проблемы с суставами, замедление роста, частые бронхиты и пневмонии.
Синдром Шейе (легкая форма) — многие пациенты с такой формой мукополисахаридоза узнают о своем заболевании случайно, порой во взрослом возрасте на фоне сопутствующих проблем со здоровьем (например, при нарушении зрения). Кроме того, если при синдромах Гурлер и Гурлер-Шейе болезнь имеет отличительные внешние признаки, то при синдроме Шейе они могут отсутствовать или проявляться минимально.
Продолжительность жизни при синдроме Гурлер зависит от формы заболевания, скорости его прогрессирования, своевременности начала терапии и наличия осложнений. Но благотворительные и медицинские сообщества подчеркивают, что особое значение имеет не только длительность, но и качество жизни ребенка и всей семьи.
При синдроме Гурлер прогрессирование может быть стремительным. Дети с тяжелой формой МПС I без лечения, как правило, доживают лишь до старшего школьного возраста. Легкие формы могут не влиять или незначительно влиять на продолжительность жизни.

Симптомы синдрома Гурлер
Как проявляется синдром Гурлер? Ответ на этот вопрос важен не только для специалистов, но и для родителей, которые зачастую первыми замечают признаки болезни.
Заподозрить синдром Гурлер сразу после рождения ребенка обычно не получается. Но «тревожные звоночки» появляются на первом году жизни. Однако они не всегда указывают на синдром.
- В некоторых случаях уже с первых месяцев жизни родители замечают, что животик малыша выглядит слишком большим при плохом наборе веса. На УЗИ врач видит умеренное увеличение печени.
- Пупочная грыжа в подавляющем большинстве случаев встречаются у детей без болезней обмена, но иногда, в сочетании с другими признаками, она может косвенно указывать на МПС I типа. Особенно это касается рецидивирующих грыж.
- Ребенок начинает часто и тяжело болеть респираторными инфекциями, которые плохо поддаются лечению и почти всегда переходят в бронхит или пневмонию. Накопление глюкозоаминогликанов в миндалинах, надгортаннике и трахее приводит к утолщению и сужению дыхательных путей.
- Со временем внешность ребенка приобретает специфические черты: лобные бугры выступают, у головы появляется большой объем, переносица «западает», язык увеличен и губы кажутся «полными». Медицинское сообщество называет это изменениями внешности по типу «гаргоилизма».
- Болезнь отражается и на задержке роста. Обычно рост не превышает 110 см и полностью останавливается к 2-5 годам. формируется так называемая «килевидная» деформация грудной клетки, позвоночник искривляется, диагностируется кифоз.
- Суставы становятся тугоподвижными, ребенку тяжело двигаться.
Поражение нервной системы
При синдроме Гурлер появляются и когнитивные нарушения. Ребенок может так и не начать говорить или утратить этот навык. У него снижается слух. Он позже садится и встает, в возрасте 2-4 лет при наличии этих навыков, они могут утрачиваться.
Важно: При начале своевременного лечения прогноз значительно улучшается и, хотя пока невозможно достичь полного выздоровления, терапия позволяет как можно дольше сохранить все необходимые навыки.
Внутренние органы
- Поражение внутренних органов проявляется в увеличении печени и селезенки, нарушении их работы. Дети часто страдают диареей в раннем возрасте и плохо набирают вес.
- Болезнь влияет на сердечные клапаны и мышечную ткань. Из-за отложения глюкозамингликанов с раннего возраста клапаны сердца утолщаются, а в некоторых случаях формируется гипертрофия мышечной ткани сердца, она становится менее эластичной и соответственно хуже выполняет свою функцию. Самое грозное осложнение — кардиомиопатия.
- Страдают органы дыхания: у людей с синдромом Гурлер узкие носовые ходы, большой язык, изменения в тканях трахеи и бронхов — все это создает условия для развития воспалительных заболеваний или апноэ.
Зрение, слух и кожа
- При МПС I типа страдает роговица, превращаясь в «матовое стекло». Снижается зрение. Со временем изменения могут привести к глаукоме.
- На поздних стадиях заболевания появляется снижение слуха, во многом это связано с частыми отитами.
- Скопление глюкозаминогликанов делает кожу утолщенной, восковидной, может наблюдаться избыточно оволосение (плотный волосяной покров на спине и руках).
На что важно обратить внимание родителям
- увеличение объема живота, не соответствующее прибавке массы тела ребенка;
- рецидивирующие грыжи (чаще двусторонние);
- особенности лицевого скелета (выступающие лобные бугры, широкие скулы, запавшая переносица, полуоткрытый рот, увеличенный язык, полные губы);
- наличие костных деформаций (деформации черепа, грудной клетки, позвоночника, конечностей,нарушение осанки — кифоз, сколиоз);
- трудности подъема ребенка из положения сидя и лежа, изменение походки, неловкость мелкой моторики;
- низкий рост с диспропорцией тела;
- частые респираторные инфекции и отиты на первом году жизни;
- помутнение роговицы и снижение слуха в более позднем возрасте;
- регресс психомоторного развития после 2-х лет жизни.
Данный материал носит исключительно информационный характер и не заменяет очную консультацию врача. Отдельные симптомы, которые могут встречаться при синдроме Гурлер, не считаются специфичными только для данного заболевания и могут наблюдаться при других состояниях либо быть индивидуальной особенностью ребенка.
Диагностика синдрома Гурлер
Синдром Гурлер нельзя диагностировать только по внешним признакам. Окончательный ответ дадут специальные исследования. Основные лабораторные методы подтверждения диагноза МПСI включают:
- количественный и качественный анализ мочи на гликозаминогликаны (ГАГ)
- определение активности фермента альфа-L-идуронидазы.
- молекулярно-генетические исследования гена IDUA. Данные исследования проводятся в специализированных генетических лабораториях.
Анализ мочи на гликозаминогликаны (ГАГ). Это безопасный и простой тест. У детей с МПС I типа в моче обычно определяется повышенный уровень ГАГ. Он не дает окончательного ответа о конкретном синдроме, но обычно становится первым и важным диагностическим этапом.
Ферментный анализ — “золотой стандарт” основан наопределение активности фермента альфа-L-идуронидазы. При подозрении на мукополисахаридоз в образце крови проверяют уровень альфа-L-идуронидазы. Если она снижена в десятки раз или близка к нулю, диагноз практически не оставляет сомнений. Этот метод также незаменим для выявления носительства заболевания у родственников.
Последним и важным шагом в верификации диагноза становится проведение молекулярно-генетического исследования. Анализ гена IDUA, считается самым информативным, он позволяет выявить патогенный вариант (генетическую поломку). Такое исследование важно не только для подтверждения диагноза, но и для оценки прогноза течения заболевания, определения дальнейшей тактики наблюдения и терапии.
«Все остальные дополнительные лабораторные и инструментальные методы обследования проводятся преимущественно для оценки степени поражения органов и выявления осложнений, связанных с данным заболеванием, а не для непосредственного подтверждения диагноза», — подчеркивает педиатр Екатерина Шлимакова.
Пренатальная диагностика и планирование семьи. Если в семье уже рождался ребенок с синдромом Гурлер, родители могут провести диагностику плода во время следующей беременности, обратившись за медико-генетической консультацией. Для этого проводится исследование ворсин хориона на 8–10 неделе или амниоцентез.
Дифференциальный диагноз нужен не только для научной точности. Схожие черты лица и симптомы встречаются при других типах мукополисахаридоза (например, при синдроме Хантера) и даже при других генетических заболеваниях. Только биохимический и генетический анализ помогают точно определиться.
Лечение синдрома Гурлер
Медицина постоянно развивается и, если раньше можно было обеспечить лишь улучшение качества жизни, то сегодня появились настоящие прорывы.
Чем раньше диагностировать тяжелые формы мукополисахаридоза, тем больше шансов замедлить течение болезни. Если синдром Гурлер подтвержден до достижения ребенком 2,5 лет, рекомендуемым лечением может стать трансплантация гемопоэтических стволовых клеток. Проведение ферментной заместительной терапии (ФЗТ) рекомендовано всем пациентам с установленным диагнозом МПС I с целью замедления прогрессирования заболевания, уменьшения размеров печени и селезенки, улучшения функции сердца, снижения уровня экскретируемых глюкозаминогликанов.
Ферментозаместительная терапия (ФЗТ)
С начала 2000-х годов применяется препарат, который замещает через капельницу недостающий «рабочий» фермент. К сожалению, действующее вещество препарата не проникает через гематоэнцефалический барьер, поэтому не влияет на работу нервной системы и мозга. Но другие органы страдают значительно меньше: уменьшается объем печени и селезёнки, число ночных апноэ и болезней дыхания, дети начинают лучше двигаться. ФЗТ не всесильна и не может «отменить» уже произошедшие изменения, но позволит значительно снизить проявления синдрома Гурлер.
Трансплантация гемопоэтических стволовых клеток (ТГСК)
Трансплантация гемопоэтических стволовых клеток в настоящее время считается одним из наиболее современных и эффективных методов лечения тяжелых форм МПС I, включая синдром Гурлер. Этот метод имеет возрастные ограничения и наибольшую эффективность показывает при проведении в раннем возрасте, как правило, у детей до 2,5 лет. Это еще раз подчеркивает важность ранней диагностики заболевания, поскольку своевременное установление диагноза напрямую влияет на возможность проведения лечения и дальнейший прогноз для ребенка.
Суть метода заключается в пересадке ребенку здоровых донорских стволовых клеток, которые после приживления начинают вырабатывать недостающий фермент. Это помогает замедлить прогрессоирование заболевания, а в ряде случаев — частично компенсировать уже имеющиеся нарушения.
Как и любая трансплантация, такое лечение требует серьезной подготовки, длительной госпитализации и связано с определенными рисками, включая инфекционные осложнения, снижение иммунитета и развитие реакции «трансплантат против хозяина». Несмотря на это, во всем мире трансплантация остается одним из наиболее предпочтительных методов лечения тяжелых форм МПС I, поскольку позволяет значительно улучшить прогноз, качество и продолжительность жизни пациентов.
Одним из наиболее значимых эффектов трансплантации становится сохранение интеллектуального развития детей, у которых сложная форма болезни, предполагающая серьезные нарушения интеллекта. При проведении этой процедуры в возрасте до 2,5 лет ребенок может развиваться почти как ровесники или лишь слезка «запаздывать».
Хирургическое лечение
Генетические поломки нельзя вылечить операцией, но при синдроме Гурлер хирургическая лечение может скорректировать состояния, которые возникли на фоне основного заболевания.
Детям могут потребоваться протезирование клапанов сердца, операции на суставах для увеличения объема движений, декомпрессия нерва в запястном канале, а иногда и нейрохирургические вмешательства при развитии гидроцефалии.
Медицинская реабилитация
Для детей с мукополисахаридозом I типа (МПС I) очень важно проходить реабилитацию. Программу подбирают индивидуально, в зависимости от того, в каком состоянии находится организми когнитивные способности ребенка. Главная цель — улучшить качество жизни.
Реабилитация может включать:
- занятия с медицинским психологом,
- отдых в специализированных санаториях,
- социальную адаптацию с участием специалистов и социальных работников,
- курсы массажа,
- лечебную физкультуру при заболеваниях и травмах суставов.
Если болезнь привела к кифозу (искривлению позвоночника) или другим аномалиям, врач-травматолог-ортопед может назначить ортезы (поддерживающие приспособления). Но только при условии, что нет сильного сдавления шейного отдела позвоночника.
МПС I часто вызывает нейрокогнитивные изменения: поведенческие проблемы, трудности со сном и многие другие. В таких случаях применяют комплексный психологический подход: образовательные и поведенческие программы, упражнения на осознанность и релаксацию. Это помогает справляться со стрессом и не дает симптомам усиливаться. Такие методы полезны и для членов семьи — они помогают получить поддержку.
Генная терапия и будущее
Сегодня весь мир ждет развития генной терапии. Предполагается, что этот метод позволит «доставить» здоровую копию гена IDUA напрямую в клетки пациента, полностью устраняя причину болезни. Пока проводятся исследования, касающиеся МПС II типа, и этот метод не скоро дойдет до повсеместной практики, но он дает надежду людям, столкнувшимся с редким и сложным заболеванием.
Прогноз и качество жизни
Сколько живут с синдромом Гурлер?
Ответ на этот вопрос во многом зависит от того, насколько рано был установлен диагноз и насколько выраженными оказались проявления заболевания на момент начала лечения. Без специфической терапии заболевание имеет неблагоприятное течение и может значительно сокращать продолжительность жизни. Однако развитие современной медицины и появление новых методов лечения существенно изменили прогноз для пациентов — сегодня терапия позволяет не только увеличить продолжительность жизни, но и значительно улучшить ее качество.
Именно поэтому ранняя диагностика имеет принципиальное значение. Своевременное выявление заболевания позволяет начать лечение до развития необратимых изменений со стороны органов и тканей, что напрямую влияет на дальнейший прогноз и эффективность терапии.
Успешная трансплантация гемопоэтических стволовых клеток (ТГСК) в раннем возрасте меняет картину. Многие дети могут учиться в школе по адаптированной программе, обслуживать себя самостоятельно, и долгое время симптомы болезни их практически не беспокоят
При промежуточных и более легких формах заболевания прогноз значительно благоприятнее. Ферментозаместительная терапия (ФЗТ) позволяет многим пациентам с синдромом Шейе сохранять активный образ жизни, получать образование, профессионально реализовываться и создавать семьи.
Жизнь с синдромом Гурлер
Куда обратиться с синдромом Гурлер в России? Если вы подозреваете синдром Гурлер у ребенка, первым врачом может стать участковый педиатр. Он назначит приемы у других профильных специалистов или направит в медико-генетический центр для прохождения обследования. При подтверждении синдрома Гурлер ребенок обычно регулярно наблюдается у генетика, педиатра, кардиолога, невролога, офтальмолога, ортопеда, пульмонолога и сурдолога. Если у вас есть сомнения в диагнозе или тактике лечения, вы можете обратиться за вторым и третьим мнением.
Важную роль играет своевременное оформление инвалидности. Ребенку положены льготы, включая бесплатное санаторное лечение, обеспечение препаратами для ФЗТ, помощь с хирургической коррекцией.
Как фонд «Правмир» помогает детям с синдромом Гурлер
В России существуют благотворительные фонды и пациентские организации для людей с мукополисахаридозом. Если вы растерялись, нуждаетесь в помощи или в составлении медицинского маршрута, на помощь придет благотворительная программа «Ассистент здоровья» фонда «Правмир». Эксперты фонда вместе с вами составляют порядок действий, подсказывают, куда обратиться и поддерживают на каждом этапе.
Мы также помогаем детям и взрослым с редкими заболеваниями в тяжелой жизненной ситуации пройти реабилитацию, оплатить жизненно-важное лечение и генетические анализы в сложных диагностических случаях.
Что может помочь оплатить фонд:
- Молекулярно-генетическое исследование.
- Программы реабилитации и средства ухода, жизненно важное лечение которые не входят в систему ОМС, но рекомендованы врачами и повышают качество жизни.
Как подать заявку на помощь? Чтобы подать заявку на помощь, надо ознакомиться с перечнем документов на официальном сайте фонда, заполнить анкету и прикрепить медицинские выписки. Специалисты фонда свяжутся с семьей, чтобы уточнить детали. Фонд регулярно публикует отчеты, чтобы помощь была прозрачной для тех, кто ее получает и оказывает.
Синдром Гурлер — это один из тех диагнозов, который в первые дни после оглашения может вызвать растерянность и страх. Отрицание, чувство одиночества, бесконечные вопросы «почему именно мы» — через это проходят почти все семьи. И это нормально. Болезнь действительно требует от семьи терпения и ежедневного мужества. Но ни одна болезнь не определяет всю жизнь ребенка. Диагноз — это лишь часть истории. За сложной медицинской терминологией, анализами, больницами и процедурами стоит человек, который при любой форме болезни любит свою семью, нуждается в поддержке близких, принятии и тепле.
Современные методы лечения способны на то, что ещё тридцать лет назад при синдроме Гурлер казалось фантастикой. Физические ограничения могут оставаться, но болезнь не способна отнять у вашего близкого любовь и радость жизни.
Если Вы чувствуете, что Вам сложно справляться с ситуацией самостоятельно, Вы всегда можете обратиться за поддержкой в программу «Ассистент здоровья» фонда «Правмир». Специалисты медицинской и психологической службы готовы помочь Вам и Вашему ребенку пройти этот непростой путь, оказать психологическую поддержку, разъяснить дальнейшие этапы обследования и лечения, а также помочь с медицинской навигацией.
Часто задаваемые вопросы
Что такое синдром Гурлер простыми словами?
Это наследственная болезнь накопления из группы мукополисахаридозов, при которой в клетках из-за нехватки определенного фермента скапливаются вещества, нарушающие работу многих органов и систем.
Как передается синдром Гурлер?
По аутосомно-рецессивному типу. Это значит, что ребенок заболевает, если «получит» генетическую поломку от обоих родителей. Человек может оставаться бессимптомным носителем данного заболевания.
Каковы первые признаки у ребёнка?
На первому году жизни ребенка родители замечают частые затяжные простуды, возникающие вновь ,даже после операции, пупочные и паховые грыжи, увеличение живота и постепенные характерные изменения внешности.
Можно ли вылечить синдром Гурлер?
Полностью вылечить болезнь пока нельзя, но современные методы (ТГСК и ФЗТ) позволяют взять ее под контроль. При ранней трансплантации стволовых клеток возможно улучшить качество и увеличить продолжительность жизни.
Сколько живут дети с синдромом Гурлер?
Без специфического лечения продолжительность жизни при тяжелых формах заболевания нередко ограничивается старшим школьным возрастом. Однако ранняя постановка диагноза и своевременное начало патогенетической терапии — трансплантации гемопоэтических стволовых клеток (ТГСК) или ферментозаместительной терапии — позволяют значительно улучшить прогноз и увеличить продолжительность жизни пациентов.
При более легких формах мукополисахаридоза прогноз в отношении продолжительности и качества жизни, как правило, более благоприятный.
Чем синдром Гурлер отличается от синдрома Шейе?
Синдром Гурлер — самая тяжелая форма МПС I типа с ранним началом и самыми серьезными проявлениями. Синдром Шейе — самая легкая форма, при которой интеллект не страдает, и пациенты могут долгое время жить нормальной жизнью.
Как получить помощь в России?
При подозрении или установленном синдроме Гурлер стоит обратиться к генетику, встать на учет в медико-генетический центр, подать документы на установление инвалидность и прохождение ПМПК, вступить в пациентское сообщество и при необходимости обратиться за поддержкой в профильные благотворительные фонды.
Если Вы столкнулись со сложностями в медицинской навигации или не понимаете дальнейший алгоритм действий, Вы можете обратиться в программу «Ассистент здоровья» фонда «Правмир». Специалисты медицинской и юридической службы помогут сориентироваться в возможностях получения медицинской помощи, разъяснят Ваши права и постараются помочь в дальнейшем маршруте .
Медицинский консультант: координатор программы Ассистент Здоровья фонда «Правмир», врач общей практики, педиатр и эндокринолог Екатерина Шлимакова.