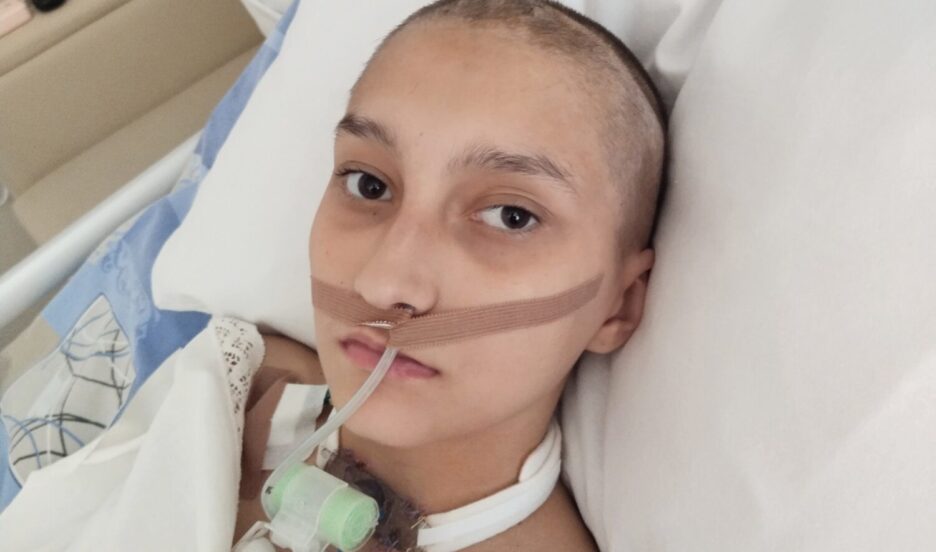
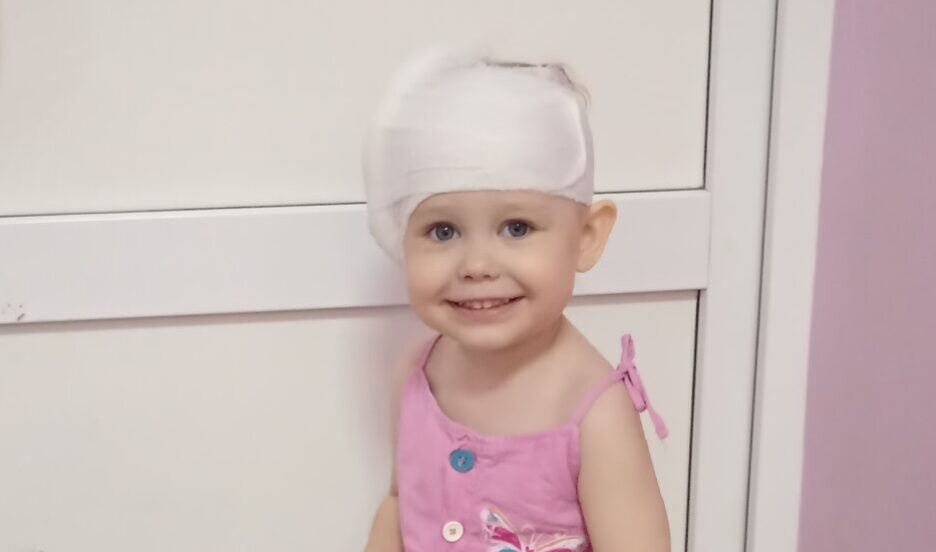
Сейчас замена препарата Пульмозим на «биоаналог» произошла по всей стране, а с 2022 года Пульмозим прекратят производить на территории РФ. Многие пациенты при переходе с одного препарата на другой стали сообщать о нежелательных явлениях.
Представители Минздрава заявляют, что жалобы пациентов только на словах и ни одного извещения они не получили.
Но им не сообщается о том, что на обращение родителей зарегистрировать нежелательную реакцию, они получают отказы или требование о госпитализации ребенка и еще раз протестировать на ребенке Тигеразу и это все в условиях пандемии, хотя именно больным с муковисцидозом рекомендовано отказаться от плановых госпитализаций и посещений медицинских учреждений.
Что же будет дальше, если «Пульмозим» прекратит производство на территории РФ?
В таком случае, данный препарат будет закупаться как незарегистрированный препарат, но только в тех случаях, когда он назначен по жизненным показаниям или индивидуальной непереносимости. Для этого также установлена процедура, которую вы можете посмотреть в разделе юридического раздела фонда «Правмир».
Если у пациента наблюдается побочная реакция, то по закону предусмотрен специальный механизм для регистрации побочных явлений и механизм обеспечения необходимым препаратом.
Важно!
- При побочных явлениях необходимо фиксировать видео-фото сьемкой и сразу же вызывать врача или скорую, необходима фиксация медицинским работником в медицинскую карту о состоянии пациента, длительности приема препарата, а также рекомендации по замене или отмене препарата.
- Оформляется врачом извещение о нежелательной реакции или отсутствии терапевтического эффекта или индивидуальной непереносимости лекарственного препарата в соответствии с Порядком осуществления фармаконадзора, утв. Приказом Росздравнадзора от 15.02.2017 № 1071
- Желательно пациенту подать заявление об оформлении извещения нежелательной реакции и о проведении врачебной комиссии для назначения препарата по жизненным показаниям.
Либо пациент может самостоятельно зафиксировать нежелательную реакцию на лекарственное средство и направить извещение в Росздравнадзор для внесения в подсистему «Фармаконадзор» Автоматизированной информационной системы Росздравнадзора и зарегистрировать в форме извещения.
Индивидуальная непереносимость является одним из двух оснований для назначения врачебной комиссией лекарственного препарата по торговому наименованию. На это указано в:
- абз. 2 п. 6 Порядка назначения лекарственных препаратов, утв. Приказом Минздрава России от 14.01.2019 № 4н;
- п. 4.7 Порядка создания и деятельности врачебной комиссии медицинской организации, утв. Приказом Минздравсоцразвития России от 05.05.2012 № 502н.
Другим основанием для назначения лекарственного препарата по торговому наименованию являются жизненные показания пациента. На это также указано в нормах Порядка.
Так, согласно Клиническим рекомендациям Минздрава России «Кистозный фиброз (муковисцидоз)»[1] «жизненными показаниями при муковисцидозе являются жизнеугрожающие состояния (мекониевый илеус, синдром потери соли, легочное кровотечение, пневмоторакс, белково-энергетическая недостаточность 3 ст, ДН (дыхательная недостаточность) и ЛСН (легочно-сердечная недостаточность) любой степени, а также состояния, отсутствие адекватной терапии которых влечет за собой уменьшение продолжительности жизни пациентов, а именно, внешнесекреторная панкреатическая недостаточность, первичный высев (выявление), интермиттирующая и хроническая инфекция легких, билиарный цирроз печени, муковисцидоз-ассоциированный сахарный диабет (инсулинозависимый). При наличии выше указанных «жизненных показаний» по решению врачебной комиссии могут быть назначены препараты по торговому наименованию».
Решением Совета Евразийской экономической комиссии от 03.11.2016 № 87 утверждены Правила надлежащей практики фармаконадзора Евразийского экономического союза. Россия входит в этот Союз.
Раздел 7 Правил устанавливает требования к организации «работы с информацией о нежелательных реакциях на лекарственные препараты». Из п. 7.1 данного раздела следует, что эта работа организуется посредством «сбора, регистрации и представления сообщений о подозреваемых нежелательных реакциях». В п. 7.1.1 также предписано «уполномоченным органам государств-членов и держателям регистрационных удостоверений … принимать соответствующие меры, чтобы проводить сбор и упорядочение сообщений о подозреваемых нежелательных реакциях, связываемых с применением лекарственных препаратов, полученных из различных источников без предварительного запроса и поступивших по запросу».
Эта работа преследует цель «обеспечения возможности сбора достаточного количества сообщений о нежелательных реакциях и их последующей научно обоснованной оценки». Таким образом Поликлиника должна зафиксировать не установленную, а подозреваемую нежелательную реакцию.
Предоставленные заявителем и собранная врачом информация о нежелательной реакции достаточны для возникновения подозрений. Устранены эти подозрения должны быть за счёт последующей их научно обоснованной оценки.
согласно п. 7.1.3.3 Правил, «если медицинский работник подтвердил (полностью или частично) случай нежелательной реакции, исходное сообщение по которому представлено потребителем или пациентом, следует точно отразить данную информацию в индивидуальном сообщении о нежелательной реакции».
В случае если для фиксации побочной реакции поликлиника предлагает «углубленное обследование» пациента в процессе приёма лекарственного препарата для того, чтобы под контролем врачей убедиться в его безопасности и переносимости для пациента. Такое «углубленное обследование» пациента является элементом клинического исследования. Это следует из п. 3 и п. 4 ч. 1 ст. 38 Закона об обращении лекарственных средств. В нём указано, что «установление безопасности лекарственного препарата и его эффективности для пациентов с определённым заболеванием», выявление «побочных действий зарегистрированных лекарственных препаратов» являются целью проведения клинических исследований.
Последние же могут осуществляться только с разрешения Минздрава России (ст. 39 Закона об обращении лекарственных средств).
Назначение лекарственного препарата по торговому наименованию не предполагает проведение таких обследований лиц, которые сообщили о появлении у них нежелательной реакции на лекарственный препарат. Нет указания на такой инструмент «валидации» данных из сообщений о нежелательных реакциях и в вышеуказанных Правилах надлежащей практики фармаконадзора Евразийского экономического союза.
В случае получения отказа от поликлиники в отправлении извещения о нежелательной реакции и назначении необходимого препарата, данный отказ следует обжаловать в Прокуратуре или суде. Для более подробной консультации или составления документов вы можете обратиться в службу юридической помощи фонда «Правмир».